Plataforma de Gestión de Laboratorio Genético conforme a ISO 17025:2017
Integración completa del Sistema de Gestión de Información de Laboratorio (LIMS)
con los requisitos de la norma ISO/IEC 17025:2017 para laboratorios de ensayo y calibración.
Este manual establece los procedimientos operativos estándar para el uso de la plataforma
Lab Genética LIMS en conformidad con la norma ISO/IEC 17025:2017
— Requisitos generales para la competencia de los laboratorios de ensayo y calibración.
Todo el personal del laboratorio debe conocer y aplicar los procedimientos aquí descritos.
1.2 Alcance
El manual cubre la operativa completa del laboratorio de genética: desde la recepción de muestras
hasta la emisión de informes, incluyendo la gestión de equipos, reactivos, control de calidad,
ensayos de intercomparación, métodos, personal, no conformidades y auditoría interna.
1.3 ¿Qué es ISO 17025:2017?
La norma ISO/IEC 17025:2017 especifica los requisitos generales para la competencia, imparcialidad
y operación consistente de los laboratorios. Se estructura en 8 capítulos:
4. Requisitos generales — imparcialidad y confidencialidad
6. Requisitos de recursos — personal (§6.2), instalaciones (§6.3), equipos (§6.4), trazabilidad metrológica (§6.5), productos y servicios externos (§6.6)
7. Requisitos del proceso — revisión de solicitudes (§7.1), métodos (§7.2), muestreo (§7.3), manipulación de ítems (§7.4), registros técnicos (§7.5), incertidumbre (§7.6), validez de resultados (§7.7), informes (§7.8), quejas (§7.9), trabajo no conforme (§7.10), control de datos (§7.11)
8. Requisitos del sistema de gestión — documentación (§8.2-8.3), acciones (§8.5-8.7), auditorías (§8.8), revisión por dirección (§8.9)
✓ El LIMS implementa automáticamente estos requisitos
Cada módulo de la plataforma está diseñado para cumplir con cláusulas específicas de la norma.
Este manual explica cómo usar cada módulo para garantizar la conformidad.
1.4 Principios ALCOA+
Todos los datos registrados en el LIMS deben cumplir los principios ALCOA+:
Principio
Significado
Cómo se cumple en el LIMS
Attributable
Atribuible a una persona
Cada acción queda registrada con usuario, IP y timestamp
Legible
Legible y permanente
Registros en base de datos, vistas web legibles
Contemporaneous
Registrado en el momento
Campos created_at/updated_at automáticos
Original
Registro original o copia certificada
Audit trail inmutable registra valores originales
Accurate
Exacto y sin errores
Validaciones de formularios, tipos de datos, FKs
+ Complete
Completo
Campos obligatorios, relaciones entre entidades
+ Consistent
Consistente
Estados de workflow secuenciales, reglas de negocio
+ Enduring
Perdurable
Base de datos MariaDB con backups programados
+ Available
Disponible
Interfaz web accesible 24/7 desde la red del laboratorio
2. Acceso al Sistema y Roles de Usuario ISO 17025 §5.5, §7.11
2.1 Inicio de Sesión
Acceda a https://genetica.omicalabs.es/login. Introduzca su nombre de usuario
y contraseña. El sistema valida las credenciales contra la base de datos de personal y registra
automáticamente la fecha y hora de acceso.
Figura 2.1 — Pantalla de inicio de sesión del LIMS
⚠ No comparta sus credenciales
Cada usuario tiene una cuenta individual. El sistema registra todas las acciones con su identidad.
Compartir credenciales viola el principio de atribución (ALCOA+) y la norma ISO 17025 §7.11.
2.2 Roles y Permisos
El sistema implementa control de acceso basado en roles (RBAC):
Rol
Permisos
Uso típico
Admin
Acceso total: todas las secciones, gestión de usuarios, configuración
Director técnico, responsable de calidad
Analyst
Muestras, placas, runs, QC, equipos, reactivos, informes (crear y editar)
Técnico de laboratorio, analista
Reviewer
Lectura de todo + revisión de resultados, QC, informes (aprobar/rechazar)
Supervisor, revisor técnico
Viewer
Solo lectura de todas las secciones (sin modificar)
Auditor externo, personal en formación
2.3 Cierre de Sesión
Haga clic en Salir en la barra de navegación superior al terminar su sesión.
El sistema destruye la sesión y requiere nueva autenticación para acceder.
2.4 Gestión de Usuarios (solo Admin)
Acceda a Personal en la barra de navegación. Aquí puede:
Crear nuevos usuarios con nombre, email, rol y cualificaciones
Asignar roles según las responsabilidades del puesto
Registrar formaciones y competencias por persona (ver Capítulo 13)
Desactivar cuentas de personal que ya no pertenece al laboratorio
ISO 17025 §6.2.2
El laboratorio debe mantener registros de competencia para todo el personal. La sección
"Personal" del LIMS sirve como registro documentado de cualificaciones, formaciones y
evaluaciones de competencia.
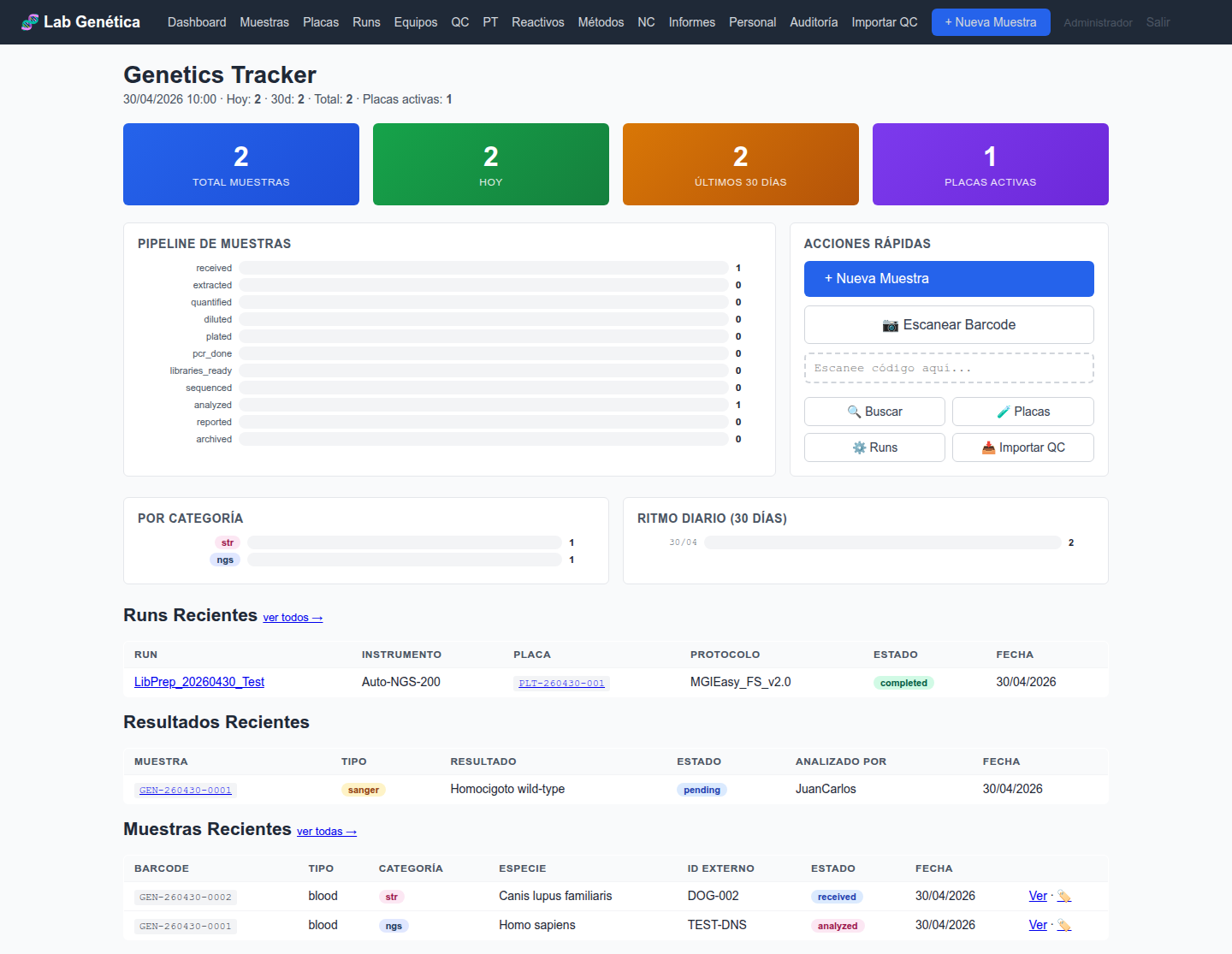
3. Gestión de Muestras: Ciclo de Vida Completo ISO 17025 §7.3, §7.4
3.1 Workflow de 11 Estados
Cada muestra en el LIMS sigue un flujo de trabajo estandarizado de 11 estados, desde la recepción
hasta el archivado. Este workflow garantiza la trazabilidad completa exigida por ISO 17025 §7.4.
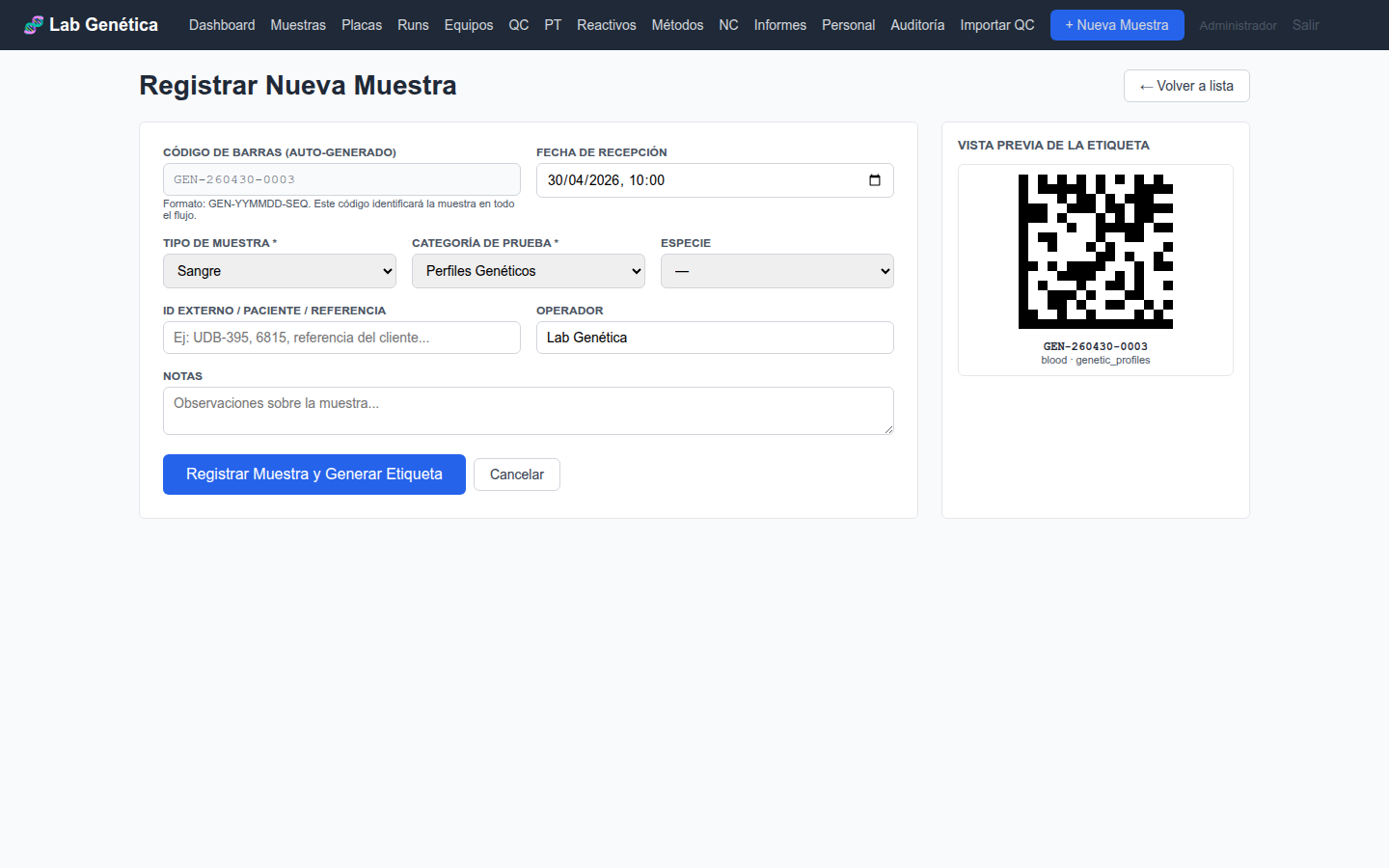
Acceda a Muestras → + Nueva Muestra y complete el formulario:
Barcode: Código único de la muestra (escaneable con lector DataMatrix)
Patient Name: Nombre del paciente o identificador del estudio
Sample Type: Tipo de muestra (sangre, saliva, tejido, ADN extraído, FFPE)
Species: Especie (humano, canino, felino, etc.)
Test Category: Categoría del ensayo (genetic_profiles, veterinary, sanger, str, ngs, pcr, other)
Priority: Prioridad (routine, urgent, stat)
Figura 3.1 — Formulario de registro de nueva muestra con código de barras
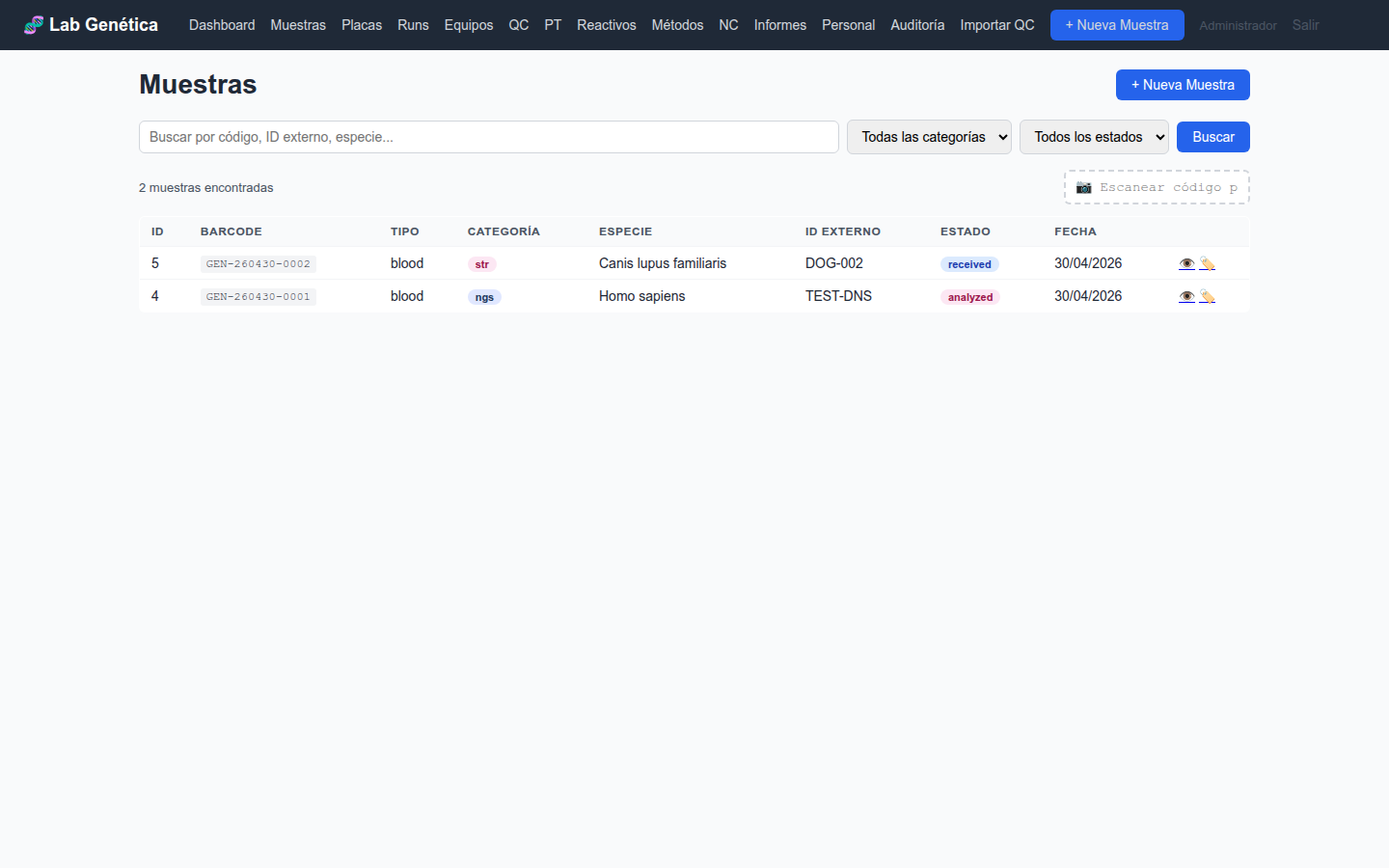
3.3 Panel de Muestras
La vista Muestras muestra el listado completo con filtros por estado, tipo
de ensayo y búsqueda por barcode. Para cada muestra se visualiza: barcode, nombre del paciente,
especie, tipo de ensayo, estado actual y timestamps de creación/actualización.
Figura 3.2 — Listado de muestras con filtros y estados del workflow
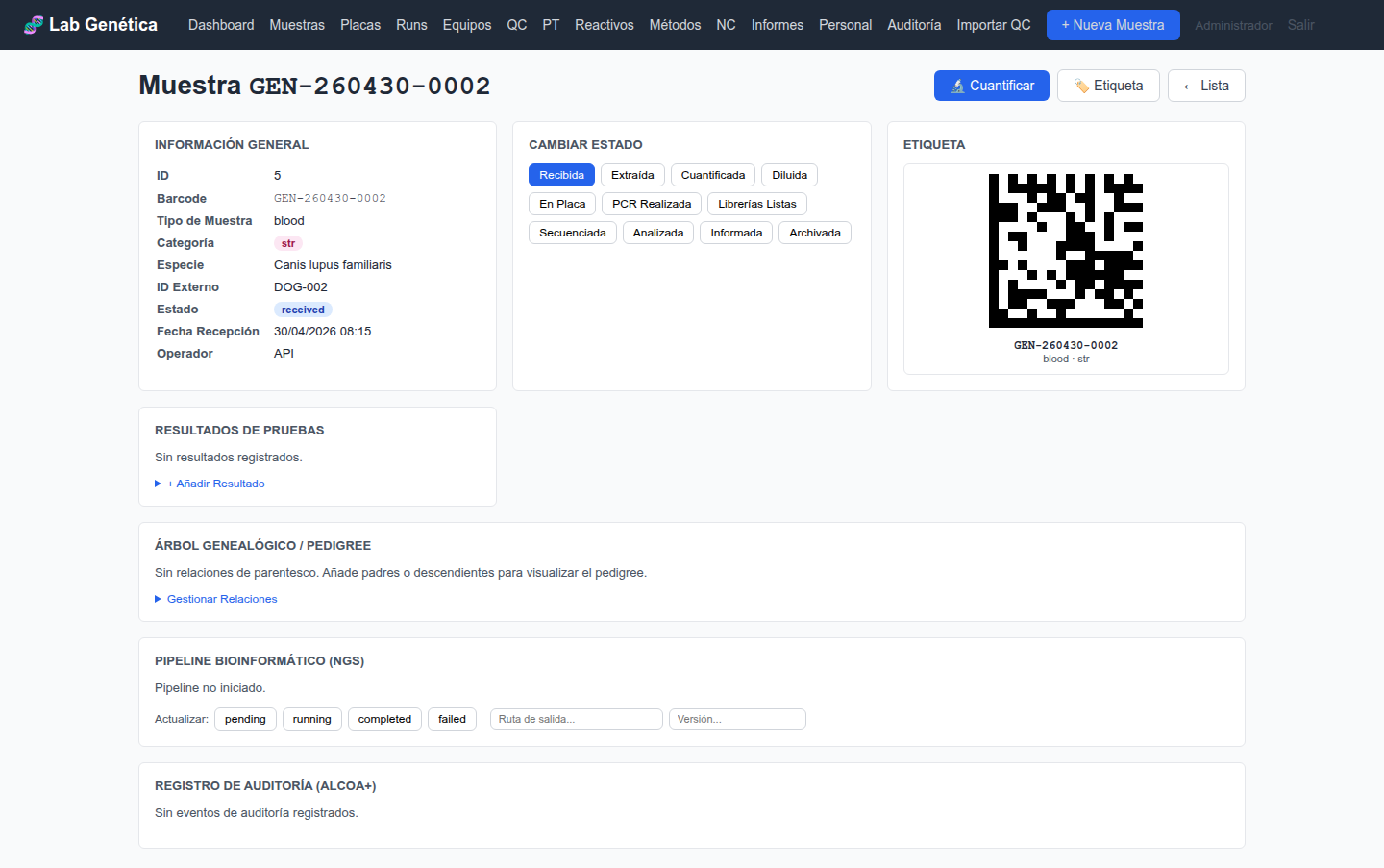
3.4 Ficha de Muestra (Vista Detallada)
Al hacer clic en una muestra, accede a su ficha completa que incluye:
Datos generales: barcode, paciente, tipo, especie, prioridad, notas
Cuantificaciones: registros de Nanodrop, Qubit, TapeStation
Diluciones: diluciones calculadas y registradas
Resultados: resultados de análisis vinculados a la muestra
Linaje: relaciones padre/hijo entre muestras (ej. ADN extraído de sangre)
Pipeline NGS: estado del pipeline, ruta de salida, versión
Trazabilidad: historial completo de auditoría de la muestra
Figura 3.3 — Ficha detallada de muestra con resultados, pipeline y auditoría
3.5 Cambio de Estado
Los botones de cambio de estado en la ficha de muestra permiten avanzar secuencialmente en el workflow. Cada cambio de estado:
Actualiza el campo status en la base de datos
Genera un registro en la auditoría (quién, cuándo, desde qué IP)
Actualiza el timestamp updated_at
⚠ No se salte estados del workflow
Cada estado representa una fase del proceso. Saltarse estados rompe la trazabilidad y puede
generar no conformidades en una auditoría ISO 17025.
3.6 Cuantificación y Dilución
Desde la ficha de la muestra, use los botones Cuantificar y Diluir
para registrar:
Concentración de ADN/ARN (ng/μL)
Relaciones de pureza (260/280, 260/230)
Volumen inicial y volumen final tras dilución
Instrumento utilizado para la medición
El sistema calcula automáticamente las diluciones necesarias según la concentración objetivo.
3.7 Resultados de Análisis
En la sección Resultados de la ficha de muestra, puede:
Añadir resultados con tipo de ensayo, resumen, detalles y archivo adjunto
Marcar el estado del resultado (pending → analyzed → reported)
Someter resultados a revisión (solo rol Reviewer)
Vincular el resultado a un método validado
4. Placas y Organización de Ensayos ISO 17025 §7.10
4.1 Placas de 96 Pozos
El módulo Placas permite organizar muestras en placas de 96 pocillos
estándar (formato A-H × 1-12) para extracción, dilución, PCR, preparación de librerías
o secuenciación. Cada pocillo
puede asignarse a una muestra, permitiendo trazabilidad espacial completa.
Figura 4.1 — Listado de placas con tipo, formato y número de pocillos asignados
4.2 Procedimiento de Plaqueado
Acceda a Placas → + Nueva Placa
Asigne un nombre descriptivo (ej. "PCR-STR-2026-04-30")
Seleccione el tipo de placa: extraction, dilution, pcr, library_prep, sequencing, other
En la cuadrícula de 96 pocillos, asigne cada muestra a su pocillo correspondiente
Cambie el estado de la placa según progrese: planned → in_progress → completed
ISO 17025 §7.10.1
La asignación de muestras a pocillos documenta inequívocamente qué muestra se procesó en
cada posición física, cumpliendo con el requisito de trazabilidad espacial.
4.3 Códigos de Barras DataMatrix
Cada placa y muestra puede generar un código de barras DataMatrix 2D compatible con
lectores automáticos (Tecan, Hamilton, etc.), eliminando errores de transcripción manual.
5. Runs de Instrumentos y Pipeline NGS
5.1 Registro de Runs
El módulo Runs documenta cada ejecución de instrumento con:
Nombre del run (ej. "SeqStudio-2026-04-30")
Instrumento utilizado (referencia a Equipos)
Protocolo y método aplicado
Operador que ejecutó el run
Estado: pending → running → completed → failed
Resultados y archivos de salida
Notas y observaciones
Figura 5.1 — Panel de runs de instrumentos con estados y filtros
5.2 Pipeline NGS
Para muestras de secuenciación masiva (NGS), el LIMS permite registrar:
Output Path: ruta al directorio de resultados del pipeline bioinformático
Pipeline Version: versión del pipeline utilizado (ej. v3.1)
La sección de pipeline en la ficha de muestra vincula el procesamiento bioinformático
con la muestra física, garantizando la trazabilidad del análisis computacional.
⚠ Documente siempre la versión del pipeline
La actualización de pipelines bioinformáticos puede cambiar resultados. Registrar la versión
permite reproducir análisis previos — requisito ISO 17025 §7.11.3 para control de datos.
6. Gestión de Equipos ISO 17025 §6.4
6.1 Inventario de Equipos
El módulo Equipos mantiene el inventario completo de instrumentos del
laboratorio con:
Código identificador único (número de serie o etiqueta de activo)
Tipo de instrumento (Nanodrop, Qubit, TapeStation, SeqStudio, LightCycler, etc.)
Fabricante, modelo, ubicación física
Fecha de instalación
Estado operativo (active, maintenance, standby, retired)
Figura 6.1 — Inventario de equipos con avisos de calibración vencida
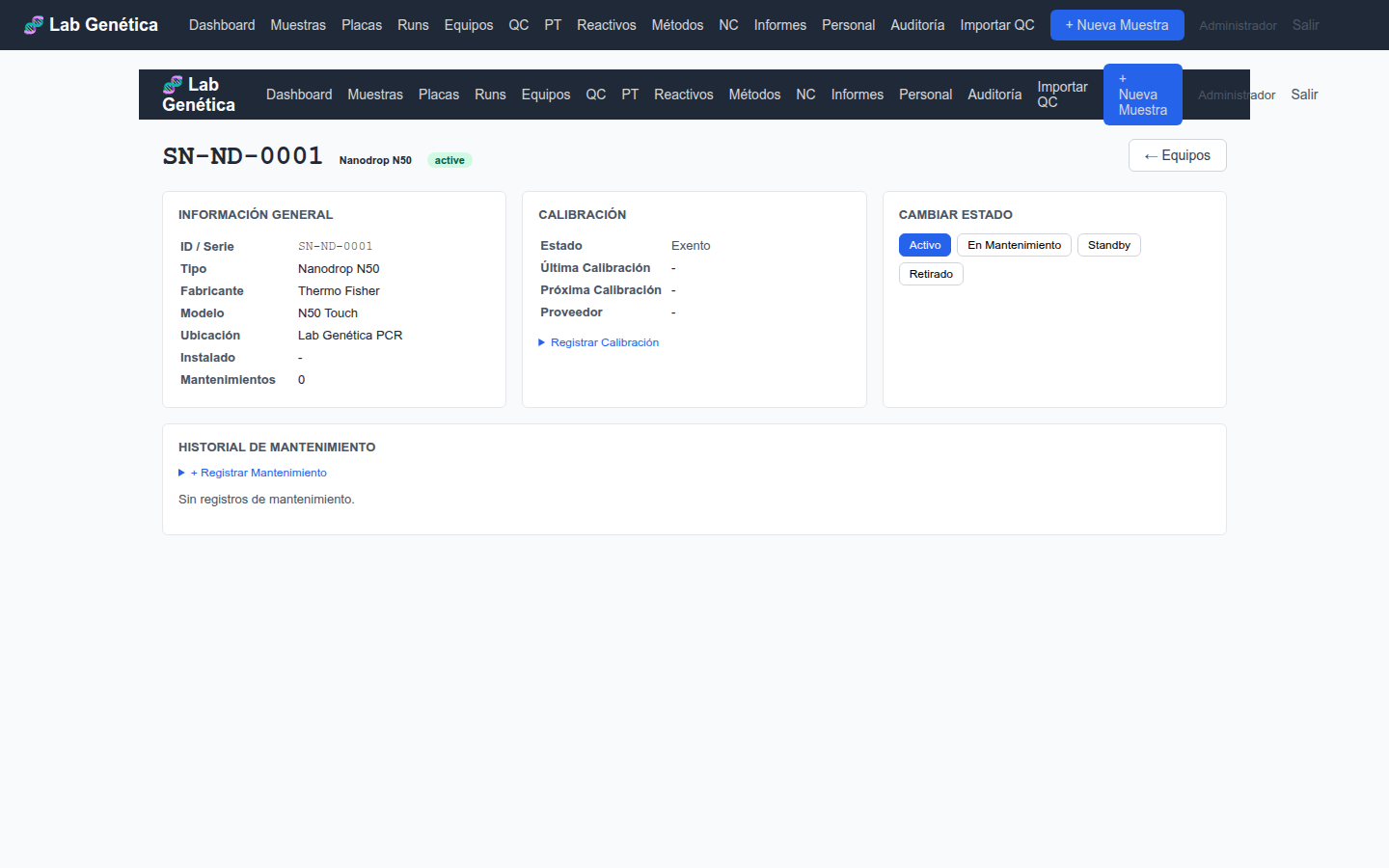
6.2 Calibración ISO 17025 §6.4.7
Cada equipo puede registrar su estado de calibración:
Fecha de última calibración
Fecha de próxima calibración
Estado: current (vigente), overdue (vencida), exempt (exento)
Proveedor de calibración
Certificado de calibración (ruta al archivo PDF)
El sistema marca automáticamente en rojo los equipos con calibración vencida en el panel principal.
⛔ NUNCA use un equipo con calibración vencida
ISO 17025 §6.4.7 exige que los equipos estén calibrados. Resultados obtenidos con equipos
no calibrados son inválidos y constituyen una no conformidad mayor.
6.3 Mantenimiento Preventivo ISO 17025 §6.4.5
El LIMS gestiona el programa de mantenimiento preventivo:
Intervalo de mantenimiento en días (ej. 180 días)
Fecha de próximo mantenimiento calculada automáticamente
Registro de mantenimientos con tipo (preventivo, correctivo, calibración, limpieza, reparación), fecha, técnico, resultado y repuestos
Acceda a la ficha de cada equipo para ver su historial completo de mantenimiento.
Figura 6.2 — Ficha de equipo con datos de calibración y mantenimiento
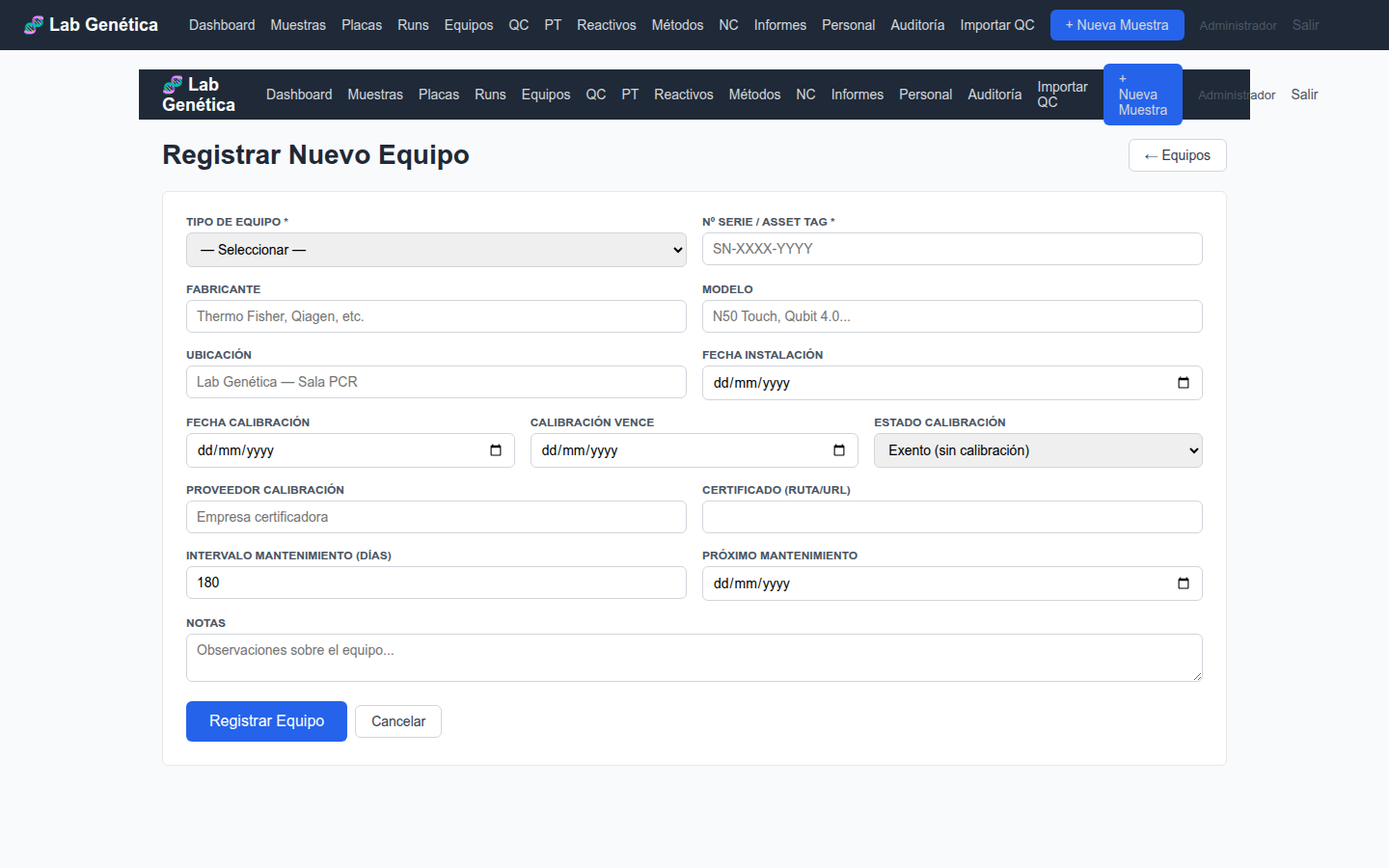
6.4 Registro de Nuevo Equipo
Figura 6.3 — Formulario de registro de nuevo equipo
6.5 Buenas Prácticas para Equipos
Registre todo equipo nuevo ANTES de ponerlo en servicio
Actualice la calibración en el sistema el mismo día que se realiza
Registre cada mantenimiento inmediatamente después de realizarlo
Adjunte el certificado de calibración (PDF) en el campo file_path
Revise semanalmente el panel de Equipos para identificar calibraciones o mantenimientos próximos a vencer
Si un equipo se retira del servicio, actualice su estado a "retired" — no lo borre
7. Reactivos y Consumibles ISO 17025 §6.6
7.1 Inventario de Reactivos
El módulo Reactivos mantiene el control de todos los reactivos y consumibles
críticos del laboratorio:
Nombre del reactivo, número de catálogo y proveedor
Número de lote (obligatorio para trazabilidad)
Fecha de caducidad
Condiciones de almacenamiento (-20°C, 4°C, temperatura ambiente)
Volumen inicial y volumen actual
Estado de QC del reactivo (pendiente, aprobado, fallido)
Figura 7.1 — Panel de reactivos con avisos de caducidad
7.2 Alertas de Caducidad
El sistema muestra automáticamente alertas en la parte superior del panel:
⚠ VENCIDO — reactivos cuya fecha de caducidad ya pasó
⏰ Próximo a vencer — reactivos que caducan en los próximos 30 días
⛔ No utilice reactivos caducados
ISO 17025 §6.6 exige que los productos y servicios externos sean aptos para su uso.
Un reactivo caducado compromete la validez de los resultados.
7.3 Trazabilidad de Lotes
Cada run de instrumento puede vincularse a los reactivos utilizados mediante la tabla
run_reagents. Esto permite:
Identificar qué lote de reactivo se usó en cada ensayo
Retrospectivamente, si se detecta un problema con un lote, localizar todos los ensayos afectados
Cumplir con el requisito ISO 17025 §6.6.2 de trazabilidad de productos externos
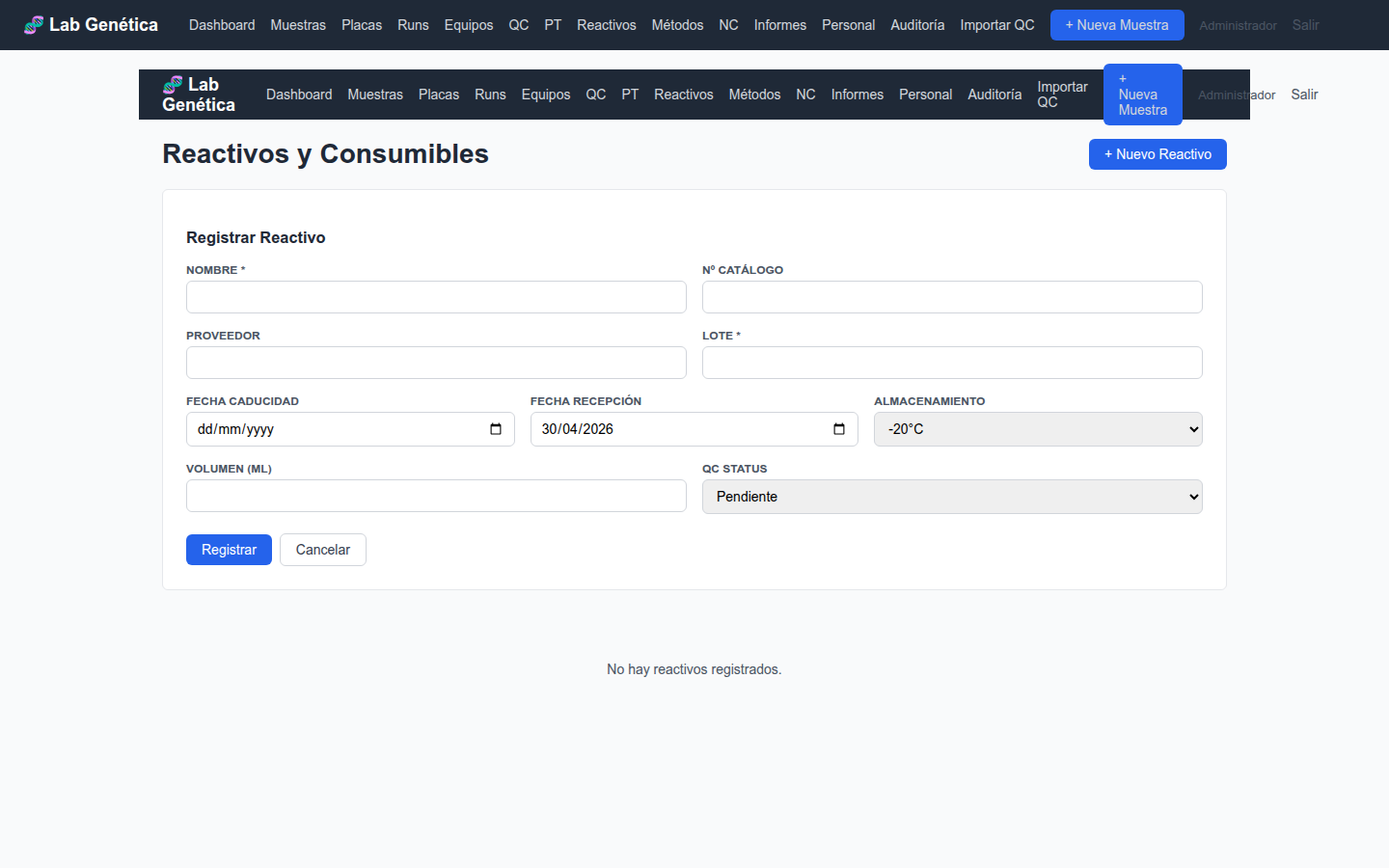
7.4 Registro de Reactivo
Figura 7.2 — Formulario de registro de nuevo reactivo
7.5 Buenas Prácticas para Reactivos
Registre todo reactivo nuevo al recibirlo, ANTES de almacenarlo
Anote siempre el número de lote — es el dato más importante para trazabilidad
Actualice el volumen restante tras cada uso significativo
Marque el QC status como "passed" solo después de verificar el rendimiento del lote
Revise el panel de Reactivos cada lunes para identificar caducidades próximas
Nunca borre un reactivo — si se agota, actualice el volumen a 0
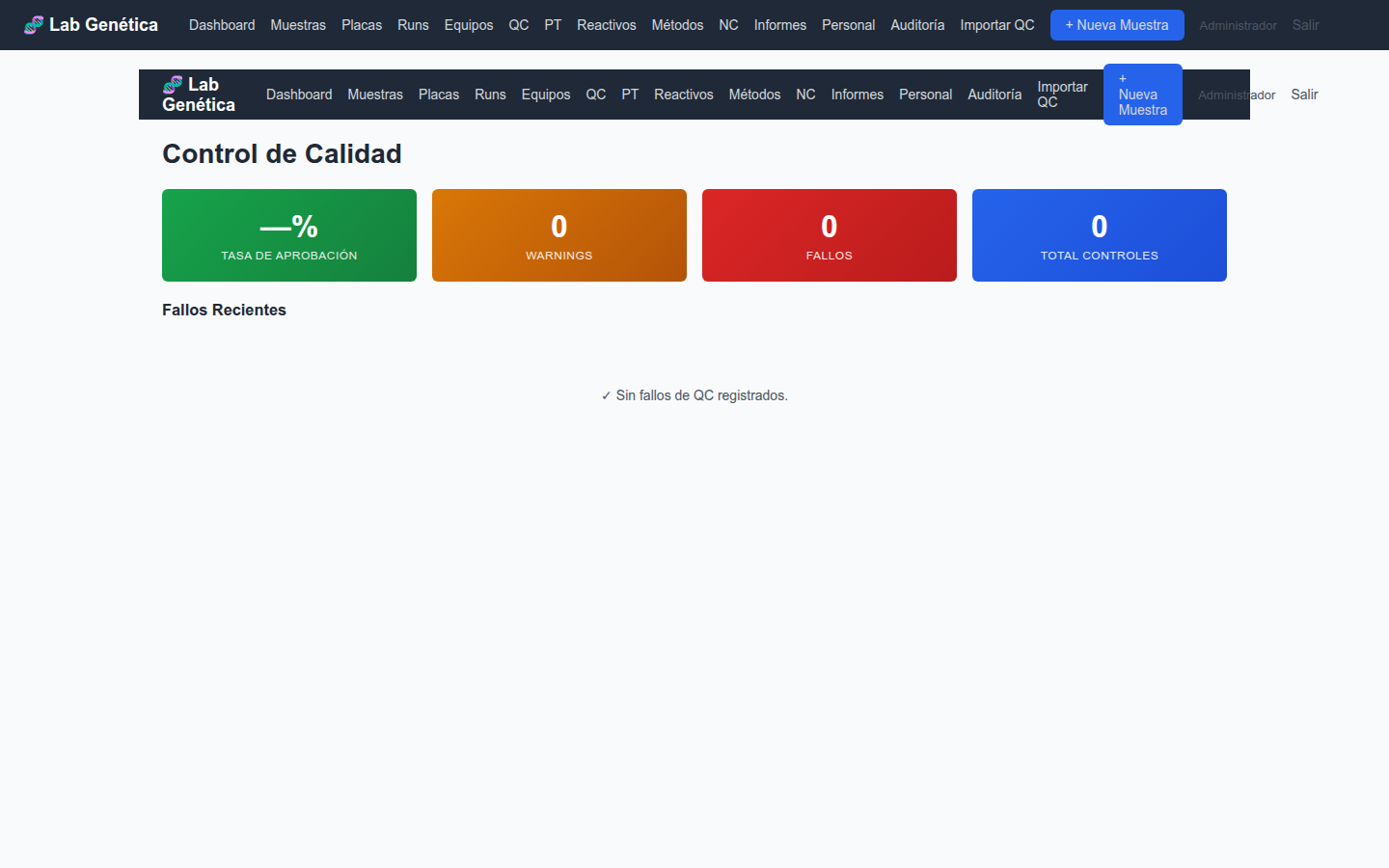
8. Control de Calidad ISO 17025 §7.7
8.1 Panel de Control de Calidad
El módulo QC centraliza la monitorización de la calidad de los resultados:
Tasa de aprobación: porcentaje de controles que pasan
Warnings y fallos: conteo de resultados fuera de especificación
Total de controles: número de mediciones de QC registradas
Desglose por parámetro: controles agrupados por parámetro QC
Figura 8.1 — Panel de control de calidad con KPIs y fallos recientes
8.2 Registros de QC
Cada resultado de QC registra:
Barcode de la muestra de control
Parámetro medido (ej. concentración, pureza 260/280, tamaño de fragmento)
Valor esperado y valor medido
Resultado: pass, warning, fail
Regla Westgard violada (si aplica)
Fecha y hora de la medición
8.3 Reglas Westgard
El LIMS soporta el registro de reglas de control Westgard para QC estadístico:
Regla
Descripción
Acción requerida
12s
1 valor > ±2 DE de la media
Warning — revisar
13s
1 valor > ±3 DE de la media
Rechazar run
22s
2 valores consecutivos > ±2 DE
Rechazar run
R4s
Rango entre 2 valores > 4 DE
Rechazar run
41s
4 valores consecutivos > ±1 DE
Rechazar run
10x
10 valores consecutivos al mismo lado de la media
Rechazar run
8.4 Revisión de Resultados
La API de QC permite a los revisores marcar resultados como revisados:
POST /api/v1/results/{id}/review con review_status y reviewed_by
La revisión queda registrada en la auditoría
9. Ensayos de Intercomparación PT/ILC ISO 17025 §7.7.2
9.1 Panel de PT
El módulo PT gestiona la participación del laboratorio en ensayos de
intercomparación (Proficiency Testing / Inter-Laboratory Comparison):
KPI Cards: conteo de resultados satisfactorios, borderline e insatisfactorios
Registro de ensayos: esquema (EMQN, UK NEQAS, CF Network), ronda, analito, método
Resultados: código de muestra, resultado enviado, consenso, Z-score, rendimiento
Figura 9.1 — Panel de ensayos de intercomparación con indicadores de rendimiento
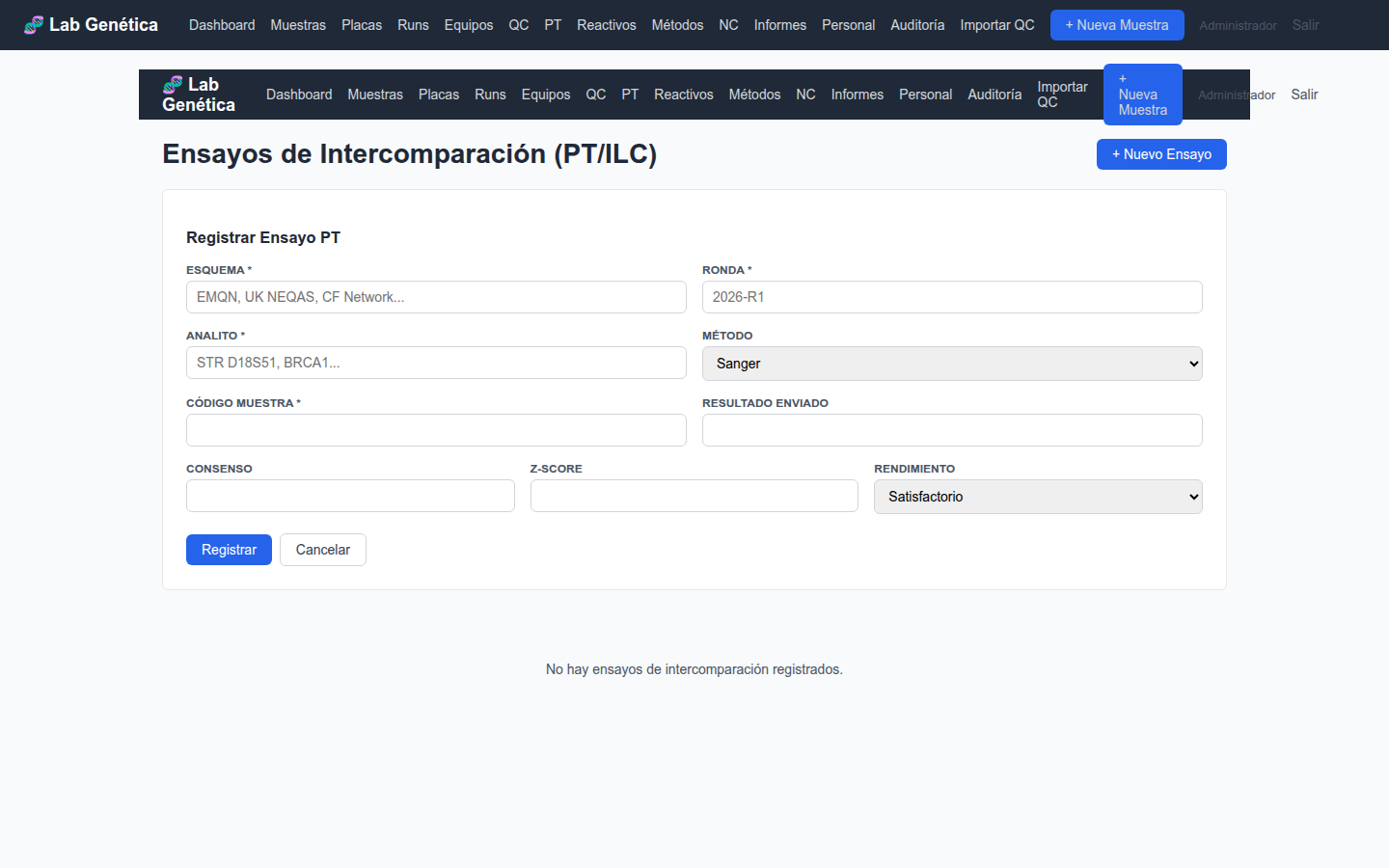
9.2 Procedimiento para Registrar un PT
Al recibir un panel de PT, acceda a PT → + Nuevo Ensayo
Registre el esquema (organizador), ronda y analito
Seleccione el método utilizado
Asigne un código de muestra único para cada ítem del panel
Tras enviar resultados al organizador, registre el resultado enviado
Cuando reciba el informe del organizador, actualice: consenso, Z-score y rendimiento
Figura 9.2 — Formulario de registro de ensayo PT
9.3 Interpretación del Z-Score
Z-Score
Interpretación
Acción
|z| ≤ 2.0
Satisfactorio
Ninguna — documentar
2.0 < |z| < 3.0
Borderline (cuestionable)
Investigar causa raíz, documentar
|z| ≥ 3.0
Insatisfactorio
Abrir No Conformidad, investigar, CAPA
⚠ Resultado insatisfactorio → NC obligatoria
Cualquier resultado de PT con rendimiento "unsatisfactory" o |Z| ≥ 3.0 debe generar una
No Conformidad (ver Capítulo 11) y un plan de acción correctiva (CAPA).
10. Métodos y Validación ISO 17025 §7.2
10.1 Catálogo de Métodos
El módulo Métodos mantiene el catálogo de métodos analíticos del laboratorio
con su estado de validación:
Código único de método (ej. SOP-EXT-001)
Nombre del método, versión y referencia al documento SOP
Tipo de ensayo (Sanger, STR, NGS, PCR, agarose gel)
Tipos de muestra compatibles
Estado de validación: not_validated → validated → verified → transferred → retired
Parámetros de rendimiento: LOD, precisión, exactitud, sensibilidad, especificidad, rango de medida
Fecha de próxima revisión
Figura 10.1 — Catálogo de métodos con alertas de revisión pendiente
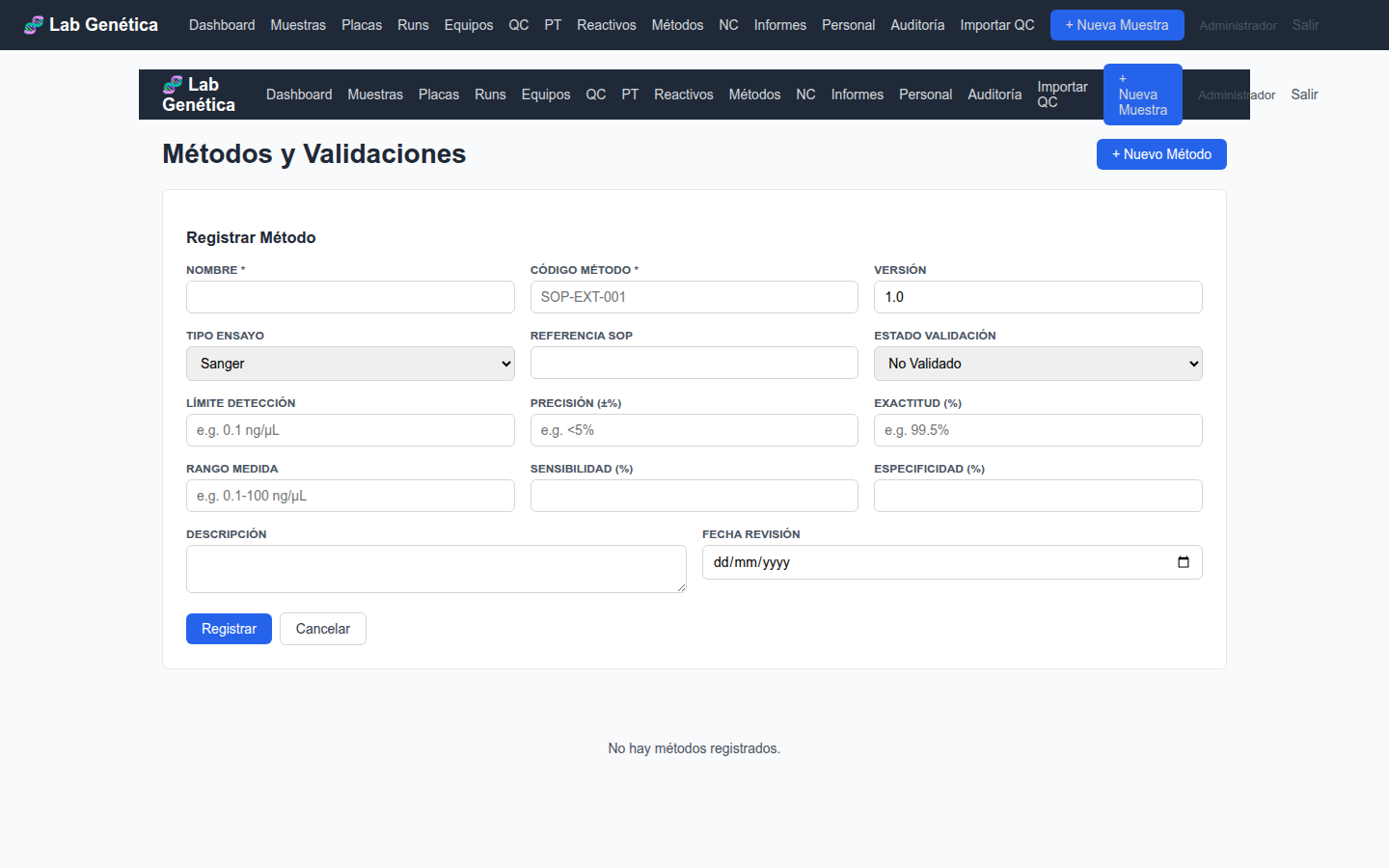
10.2 Parámetros de Validación ISO 17025 §7.2.2
Para cada método, registre los siguientes parámetros según corresponda:
Parámetro
Definición
Ejemplo
Límite de Detección (LOD)
Mínima cantidad detectable del analito
0.1 ng/μL
Precisión
Repetibilidad del método (% CV)
<5%
Exactitud
Proximidad al valor verdadero
99.5%
Sensibilidad
Capacidad de detectar el analito
98%
Especificidad
Capacidad de discriminar el analito
100%
Rango de medida
Intervalo de trabajo validado
0.1-100 ng/μL
10.3 Revisión Periódica de Métodos
El sistema alerta cuando un método tiene su fecha de revisión vencida. La revisión periódica es
obligatoria según ISO 17025 §7.2.3 para garantizar que los métodos siguen siendo aptos.
Figura 10.2 — Formulario de registro de método con parámetros de validación
10.4 Vinculación Método-Resultado
Cada resultado de análisis (sample_results) y cada run de instrumento
(instrument_runs) puede vincularse al método utilizado. Esto garantiza
que cualquier resultado sea trazable a un método validado específico.
11. No Conformidades y CAPA ISO 17025 §8.6, §8.7
11.1 Panel de No Conformidades
El módulo NC gestiona el sistema de no conformidades y acciones
correctivas/preventivas (CAPA):
KPI Cards: total de NC, abiertas, cerradas, por severidad
Clasificación por origen: muestra, ensayo, equipo, reactivo, documento, proceso
Severidad: critical, major, minor, observation
Figura 11.1 — Panel de no conformidades con indicadores y filtros
11.2 Workflow de una No Conformidad
Figura 11.2 — Flujo de estados de una No Conformidad: open → investigating → correcting → verified → closed
11.3 Procedimiento ante una No Conformidad
Identificación: Cualquier miembro del personal puede detectar una NC y registrarla en el sistema
Registro: Describa la no conformidad, clasifique su origen y severidad, y documente la acción inmediata tomada
Investigación: El responsable de calidad analiza la causa raíz (root_cause)
Corrección: Se define e implementa la acción correctiva (corrective_action)
Prevención: Se define la acción preventiva para evitar recurrencia (preventive_action)
Verificación: Una persona independiente verifica la eficacia de las acciones (verified_by, verified_date)
Cierre: La NC pasa a estado "closed" con fecha de cierre automática
Figura 11.3 — Formulario de registro de no conformidad
⚠ Toda NC crítica o mayor debe cerrarse en ≤ 30 días
ISO 17025 §8.7.1 exige acciones correctivas oportunas. Las NC que superen 30 días abiertas
deben escalarse a la dirección del laboratorio.
12. Informes y Sign-off ISO 17025 §7.8
12.1 Panel de Informes
El módulo Informes gestiona la emisión de informes de ensayo con
flujo de revisión y aprobación:
KPI Cards: total, borradores, aprobados, emitidos
Numeración automática: RPT-YYMM-NNN
Tipos de informe: análisis, QC, validación, PT, no conformidad, otro
Flujo de sign-off: draft → in_review → approved → issued (o rejected → in_review)
Figura 12.1 — Panel de informes con estadísticas y filtros
12.2 Flujo de Aprobación (Sign-off)
Figura 12.2 — Flujo de sign-off de informes: borrador → revisión → aprobación → emisión
12.3 Generación de PDF
Cada informe puede generar una vista PDF con:
Encabezado del laboratorio y número de informe
Datos de la muestra vinculada (barcode, paciente)
Resultados estructurados (campo report_data en JSON)
Identidad del emisor y aprobador
Fechas de emisión y aprobación
Figura 12.3 — Formulario de creación de informe
12.4 Buenas Prácticas para Informes
Todo informe de resultado debe estar vinculado a una muestra
El informe debe ser revisado por una persona distinta al emisor (principio de 4 ojos)
La aprobación debe registrarse en el sistema antes de la emisión al cliente
Un informe rechazado debe documentar el motivo y reenviarse a revisión
Conserve los PDFs generados en una ubicación segura y respaldada
13. Personal y Competencia ISO 17025 §6.2
13.1 Registro de Personal
El módulo Personal (accesible como admin) mantiene el registro de
todo el personal del laboratorio:
Nombre completo, username único, email
Rol en el sistema (admin, analyst, reviewer, viewer)
Cargo y departamento
Cualificaciones (títulos, certificaciones)
Fecha del último acceso al sistema
Figura 13.1 — Listado de personal del laboratorio
13.2 Formación y Competencia
La ficha de cada persona incluye dos secciones clave:
Estado de competencia resultante: competent, needs_review, not_competent
Certificado asociado (ruta al archivo)
Competencia por Alcance
Alcance de la competencia (ej. "Extracción de ADN", "Análisis de fragmentos STR")
Método asociado (si es específico de método)
Nivel: trainee, supervised, independent, trainer
Evaluador y fecha de evaluación
Fecha de próxima revisión de competencia
Figura 13.2 — Ficha de personal con formación, competencias y formularios de alta
ISO 17025 §6.2.5
El laboratorio debe tener procedimientos para evaluar la eficacia de las acciones formativas.
El campo competency_status en cada registro de formación documenta esta evaluación.
13.3 Buenas Prácticas de Gestión de Personal
Registre a cada nuevo empleado ANTES de que comience a trabajar en el laboratorio
Documente todas las formaciones, incluso las internas (ej. entrenamiento en un nuevo método)
Establezca fechas de revisión de competencia para cada alcance (recomendado: anual)
Revise mensualmente el panel de Personal para identificar formaciones próximas a expirar
Desactive (no borre) las cuentas de personal que abandona el laboratorio
14. Auditoría y Trazabilidad ALCOA+ ISO 17025 §8.8
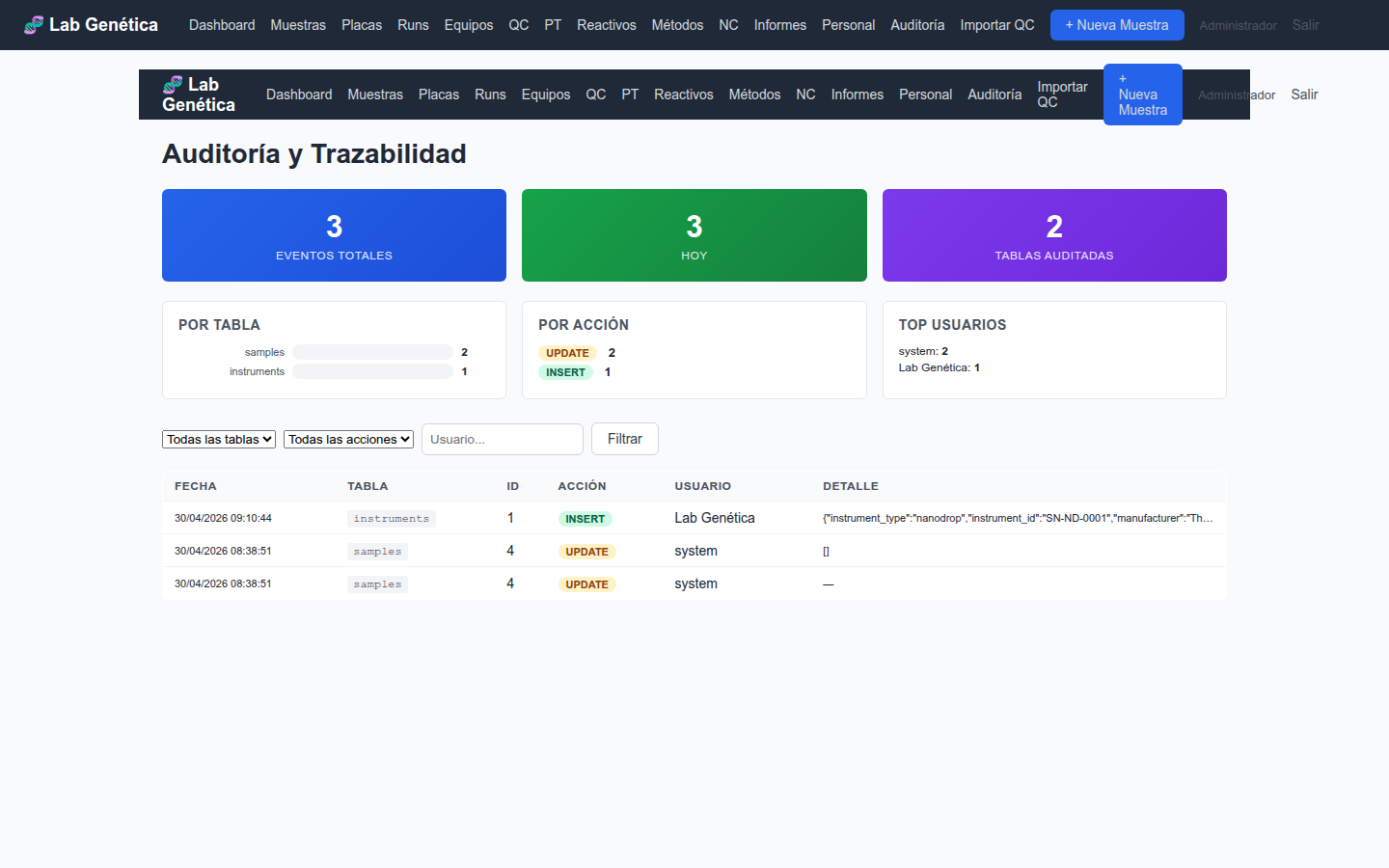
14.1 Panel de Auditoría
El módulo Auditoría proporciona visibilidad completa sobre todas las
operaciones registradas en el sistema:
KPI Cards: total de registros de auditoría, tablas auditadas, usuarios activos
Gráfico por tabla: distribución de registros de auditoría por tabla de base de datos
Gráfico por acción: distribución por tipo de acción (INSERT, UPDATE, DELETE)
Top usuarios: usuarios con más actividad registrada
Filtros: por tabla, acción, usuario, fecha
Figura 14.1 — Panel de auditoría ALCOA+ con KPIs y distribución por tablas
14.2 Qué Registra el Audit Trail
Cada operación de escritura (INSERT, UPDATE, DELETE) en cualquiera de las tablas principales del sistema genera automáticamente un registro de auditoría que captura:
Campo
Contenido
Principio ALCOA+
table_name
Tabla afectada (ej. samples, instruments, results)
Attributable
record_id
ID del registro modificado
Traceable
action
INSERT, UPDATE, DELETE
—
old_values
JSON con los valores ANTES del cambio
Original
new_values
JSON con los valores DESPUÉS del cambio
Accurate
changed_by
Usuario que realizó la acción
Attributable
ip_address
Dirección IP desde la que se realizó
Attributable
created_at
Timestamp preciso de la acción
Contemporaneous
14.3 Auditoría por Muestra
En la ficha de cada muestra, la sección Trazabilidad muestra el historial
completo de cambios de esa muestra específica, permitiendo reconstruir todo su ciclo de vida.
✓ El audit trail es automático e inmutable
El personal no puede modificar ni eliminar registros de auditoría. Las eliminaciones (DELETE)
se han reemplazado por soft-deletes que marcan registros como eliminados sin perder los datos,
cumpliendo con el principio ALCOA+ de "Enduring" (perdurable).
14.4 Buenas Prácticas de Auditoría
Revise el panel de auditoría semanalmente para detectar anomalías
Investigue cualquier acceso o modificación no reconocida
Utilice el filtro por tabla para auditar áreas específicas (ej. solo results)
Durante auditorías internas o externas, use el panel para demostrar trazabilidad
Exporte los registros de auditoría para respaldo periódico
15. Diagramas de Flujo de Procesos
15.1 Flujo General del Laboratorio
Figura 15.1 — Flujo general del laboratorio desde recepción hasta informe final
15.2 Flujo de Gestión de Calidad (ISO 17025 integrado)
Figura 15.2 — Arquitectura integrada de gestión de calidad ISO 17025 en el LIMS
15.3 Ciclo de Mejora Continua (PDCA)
Figura 15.3 — Ciclo PDCA (Plan-Do-Check-Act) de mejora continua integrado con el LIMS
16. Anexos: Checklists y Referencias
16.1 Checklist Diario del Operador
Verificar el Dashboard al inicio del turno — revisar alertas y KPIs
Registrar todas las muestras recibidas en el día (antes de procesarlas)
Actualizar estados de muestras procesadas (extracción → cuantificación → dilución → placa)
Registrar runs de instrumentos completados (con protocolo, operador y estado)
Documentar cualquier incidencia como No Conformidad en el módulo NC
Cerrar sesión al finalizar el turno
16.2 Checklist Semanal del Supervisor
Revisar el panel de Equipos — ¿hay calibraciones o mantenimientos vencidos o próximos?
Revisar el panel de Reactivos — ¿hay lotes caducados o próximos a caducar?
Revisar el panel de QC — ¿hay fallos o tendencias anómalas en los parámetros?
Revisar el panel de NC — ¿hay NC abiertas > 30 días? Escalar si es necesario
Revisar el panel de Informes — ¿hay informes pendientes de revisión/aprobación?
Revisar el panel de Personal — ¿hay formaciones próximas a expirar?
Revisar el panel de Auditoría — detectar accesos o modificaciones anómalas
16.3 Checklist Mensual del Responsable de Calidad
Generar informe de NC del mes (abiertas, cerradas, tiempo medio de resolución)
Revisar resultados de PT/ILC pendientes de actualizar (Z-scores, rendimiento)
Verificar revisión de métodos — ¿hay métodos con fecha de revisión vencida?
Auditar 5 muestras aleatorias verificando trazabilidad completa en el audit trail
Revisar competencias del personal — ¿hay evaluaciones pendientes?
Actualizar este manual si hay cambios en procedimientos o nueva normativa
Preparar datos para la revisión por dirección (ISO 17025 §8.9)
16.4 Referencia Rápida de Módulos y Cláusulas ISO 17025
Módulo LIMS
URL
Cláusula ISO 17025
Qué cubre
Dashboard
/
General
Visión general del laboratorio
Muestras
/samples
§7.3, §7.4, §7.11
Workflow, trazabilidad, datos
Placas
/plates
§7.10
Organización espacial de ensayos
Runs
/runs
§7.11
Ejecución de instrumentos, pipeline
Equipos
/equipment
§6.4
Inventario, calibración, mantenimiento
QC
/qc
§7.7.1
Control de calidad, Westgard
PT
/pt
§7.7.2
Ensayos de intercomparación
Reactivos
/reagents
§6.6
Inventario, lotes, caducidad
Métodos
/methods
§7.2
Catálogo, validación, revisión
NC/CAPA
/nonconformances
§8.6, §8.7
No conformidades, acciones
Informes
/reports
§7.8
Emisión, revisión, sign-off
Personal
/personnel
§6.2
Competencia, formación, roles
Auditoría
/audit
§8.8
Audit trail ALCOA+
16.5 Tabla de Responsabilidades (Matriz RACI)
Actividad
Admin
Analyst
Reviewer
Viewer
Registro de muestras
R
R
A
I
Actualizar estado de muestra
R
R
A
I
Registro de cuantificaciones
R
R
A
I
Gestión de equipos (cal/mant)
R
C
A
I
Registro de reactivos
R
R
A
I
Ejecución de QC
R
R
A
I
Registro de PT
R
R
A
I
Registro/Apertura de NC
R
R
R
I
Investigación de NC (causa raíz)
R
C
R
I
Verificación CAPA
R
—
R
I
Revisión de resultados
R
—
R
I
Emisión de informes
R
R
A
I
Aprobación de informes
R
—
R
—
Gestión de personal (altas/bajas)
R
—
—
—
Registro de formaciones
R
—
R
I
Revisión de auditoría
R
—
R
I
Exportación de datos para auditoría
R
—
I
—
Leyenda: R = Responsible (ejecuta), A = Accountable (responsable último), C = Consulted (consultado), I = Informed (informado)
16.6 Glosario
Término
Definición
ALCOA+
Attributable, Legible, Contemporaneous, Original, Accurate + Complete, Consistent, Enduring, Available
CAPA
Corrective and Preventive Action (acción correctiva y preventiva)
Next-Generation Sequencing (secuenciación de nueva generación)
PDCA
Plan-Do-Check-Act (ciclo de mejora continua)
PT
Proficiency Testing (ensayo de aptitud / intercomparación)
RBAC
Role-Based Access Control (control de acceso basado en roles)
SOP
Standard Operating Procedure (procedimiento normalizado de trabajo)
Westgard
Reglas estadísticas de control de calidad para laboratorios clínicos
16.7 Registro de Revisiones del Manual
Versión
Fecha
Autor
Cambios
1.0
2026-04-30
Lab Genética
Versión inicial. Integración completa ISO 17025:2017 con LIMS.
— Fin del Manual —
17. Cumplimiento Adicional ISO 17025
Este capítulo aborda cláusulas de la norma que, si bien no tienen un módulo dedicado en el LIMS, deben cumplirse mediante procedimientos operativos complementarios. El LIMS proporciona la infraestructura de registro; el personal debe seguir las pautas aquí descritas.
17.1 Instalaciones y Condiciones Ambientales ISO 17025 §6.3
Las instalaciones del laboratorio deben ser apropiadas para las actividades de ensayo. El laboratorio debe:
Monitorizar condiciones ambientales: temperatura, humedad y limpieza de las áreas de trabajo. Registrar lecturas diarias en el módulo QC como controles de tipo "environmental".
Control de acceso: restringir el acceso físico a áreas de ensayo. El LIMS complementa esto con su sistema de roles RBAC.
Separación de áreas: mantener separación física entre áreas incompatibles (pre-PCR vs post-PCR). Documentar la asignación de equipos a cada área usando el campo location en el módulo de Equipos.
ISO 17025 §6.3.3
Las condiciones ambientales que puedan influir en los resultados deben registrarse. Use el módulo QC para registrar parámetros como temperatura de neveras (-20°C, -80°C), humedad del laboratorio, y limpieza de campanas de flujo laminar.
17.2 Trazabilidad Metrológica ISO 17025 §6.5
Los resultados de medición deben ser trazables a patrones de referencia reconocidos. El LIMS soporta esta trazabilidad mediante:
Cadena de calibración: cada equipo registra su proveedor de calibración y certificado. Verifique que el proveedor esté acreditado conforme a ISO 17025.
Materiales de referencia: registre los materiales de referencia certificados (CRM) como reactivos en el módulo Reactivos, marcándolos con QC status "passed" y adjuntando el certificado.
Registro de patrones: documente los patrones de referencia internos en el módulo de Métodos, en el campo notes, indicando su trazabilidad a patrones nacionales o internacionales.
⚠ Los equipos exentos de calibración deben justificarse
Si un equipo tiene calibration_status = exempt, debe documentarse en notes el motivo técnico por el cual no requiere calibración (ej. equipo auxiliar que no afecta resultados de ensayo).
17.3 Revisión de Solicitudes, Ofertas y Contratos ISO 17025 §7.1
Antes de aceptar una muestra, el laboratorio debe revisar que puede cumplir con los requisitos del cliente:
Viabilidad técnica: ¿tenemos el método validado para este ensayo? Verificar en el módulo Métodos que el método está en estado "validated" o "verified".
Capacidad operativa: ¿hay reactivos en fecha, equipos calibrados y personal competente disponible? Verificar los módulos de Reactivos, Equipos y Personal.
Documentación: registrar la revisión en el campo notes de la muestra al crearla, indicando que se ha verificado la viabilidad.
17.4 Incertidumbre de Medición ISO 17025 §7.6
El laboratorio debe evaluar la incertidumbre de medición para todos los resultados cuantitativos:
Fuentes de incertidumbre: identificar las contribuciones relevantes (instrumento, operador, ambiente, método). Documentarlas en una hoja de cálculo o registro externo.
Estimación: calcular la incertidumbre expandida (k=2, 95% de confianza) usando el método GUM (Guide to the Expression of Uncertainty in Measurement).
Registro en el LIMS: almacenar el valor de incertidumbre en el campo report_data (JSON) del informe de resultado, junto con los valores medidos y sus unidades.
Informe al cliente: incluir la incertidumbre en los informes de ensayo cuando sea relevante para la validez del resultado.
Nota práctica
Para ensayos cualitativos (ej. genotipado STR, presencia/ausencia de variante), la incertidumbre puede expresarse como el límite de detección (LOD) del método. El campo lod en el módulo de Métodos captura este dato.
17.5 Gestión de Quejas ISO 17025 §7.9
El laboratorio debe tener un procedimiento documentado para la gestión de quejas de clientes. En el LIMS:
Registro: toda queja recibida (verbal, email, escrita) debe documentarse como una No Conformidad con source = process y severidad según el impacto potencial en resultados.
Acuse de recibo: registrar en immediate_action la confirmación de recepción al cliente.
Investigación: seguir el flujo NC/CAPA estándar (Capítulo 11) para investigar, corregir y prevenir recurrencia.
Comunicación al cliente: registrar en notes de la NC el resultado de la investigación comunicado al cliente y la fecha de comunicación.
Seguimiento: si la queja revela un problema sistémico, generar una NC adicional de tipo major o critical.
17.6 Acciones para Abordar Riesgos y Oportunidades ISO 17025 §8.5
El laboratorio debe identificar riesgos y oportunidades para la mejora continua. Se recomienda:
Matriz de riesgos (externa al LIMS): mantener un documento Excel/Google Sheets con los riesgos identificados por área, su probabilidad, impacto y medidas de mitigación.
Registro en el LIMS: los riesgos materializados (incidentes) se registran como NC. Las oportunidades de mejora detectadas en auditorías o revisiones se documentan en el campo notes del informe de auditoría.
Revisión periódica: durante la revisión mensual del responsable de calidad (checklist §16.3), evaluar si han surgido nuevos riesgos o si las medidas de mitigación existentes son eficaces.
17.7 Tabla de Conformidad Completa
A continuación se presenta el mapeo completo de todas las cláusulas de la norma ISO 17025:2017 aplicables y cómo se cumplen en el sistema integrado LIMS + procedimientos:
Cláusula
Requisito
Cumplimiento
§4.1
Imparcialidad
RBAC + audit trail + segregación de roles Reviewer/Analyst
§4.2
Confidencialidad
Login individual, roles, acceso restringido
§5.5
Estructura organizativa
Roles RBAC, matriz RACI (§16.5)
§6.2
Personal
Módulo Personal + training + competencia (Cap. 13)